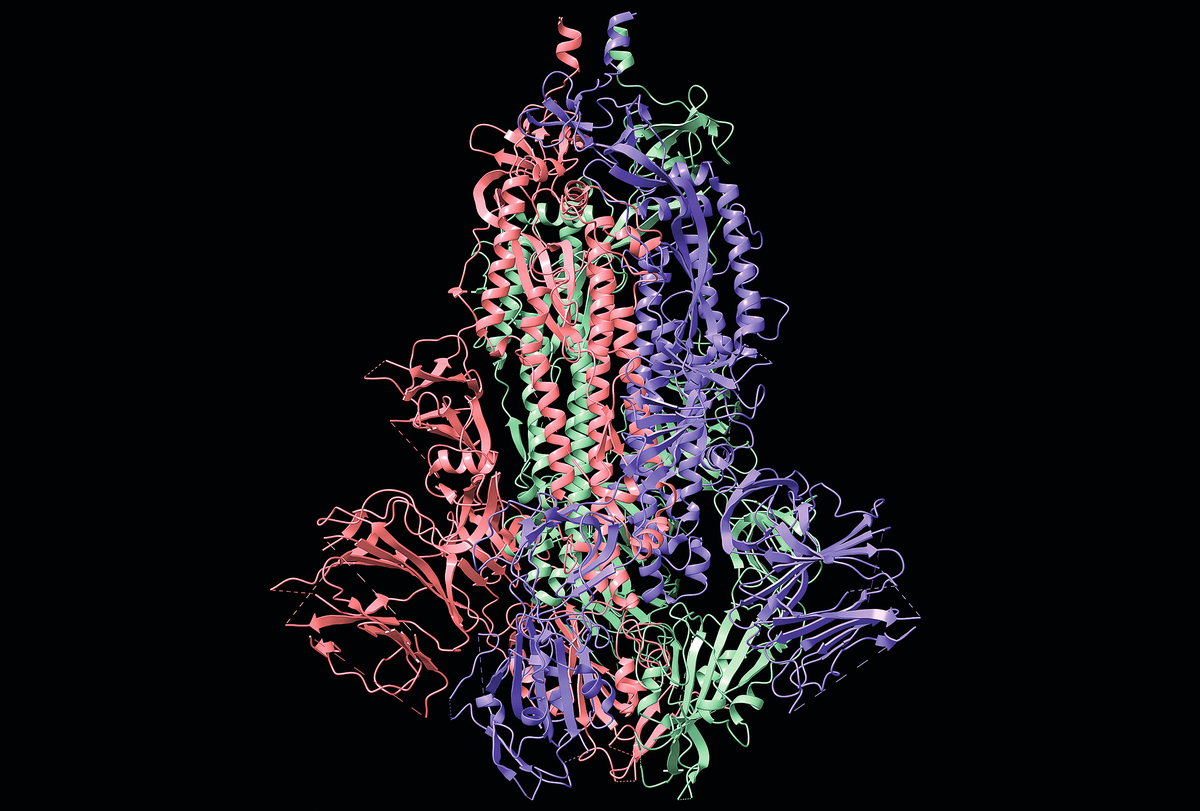
華盛頓大學(University of Washington)的一個研究組設計了一款能夠準確預測蛋白質折疊模型的軟件。以前,確定一種蛋白質的折疊構象需要幾年的時間,而使用這款軟件大約十分鐘即可得到結果。這個研究組把軟件放在網上公開供所有同行下載使用。
蛋白質折疊(protein folding)與蛋白質的功能緊密相關的因素。、帕金森氏病、阿茲海默症(Alzheimer』s)等現在無法治療的疾病都與蛋白質出錯相關。科學家希望能從化學成份準確預測蛋白質的三維構象,依此設計治療各種疾病的新蛋白質,就能解決很多與生物分子相關的高科技的棘手問題。
然而要獲得蛋白質三維構象猶如一個破譯密碼的過程,在這類軟件出現之前,研究者一般需要幾年的時間才能確定一種蛋白質的折疊構象。近年來,科學家發現利用人工智能軟件可以大幅加快研究進程。研究人員迫切需要這類能夠快速、準確地預測蛋白質折疊模型的軟件。這對攻克人類多種疾病具有重要意義。
2020 年,Google旗下人工智能公司DeepMind 推出了能夠快速解碼蛋白質折疊構象的人工智能軟件AlphaFold2,在世界蛋白質模型預測競賽中排名第一。這款軟件當時受到生物界廣泛關注,但是,該公司當時沒有開放這款軟件供同行共享。
加州大學結構生物物理學家阿加德(David Agard)評價此事說:「這(AlphaFold2 的誕生)相當令人激動, 但是也讓人沮喪。」
在那之後至今僅幾個月的時間, 華盛頓大學的研究組設計出一個完全可以與AlphaFold2形成競爭的另一款同類軟件Rose TTA Fold,並立即邀請公證們正在審議AlphaFold2 的細節,公司「將把AlphaFold 開放供科學界免費使用」。
7 月15 日,《科學》(Science)期刊發表了華盛頓大學的軟件成果。同日,《自然》(Nature)期刊也迅速發表了關於Alpha Fold的研究報告。
華盛頓大學研究組的貝克(David Baker)說:「他們的研究成果沒有落後於我們發表,這是合理的,因為我們的研究是基於他們的成果之上。」
自7 月起, 世界各地已經有140 多個獨立研究組從開源軟件資料庫GitHub 上, 下載了華盛頓大學研究組開發的Rose TTA Fold 軟件。
合作研究員之一華盛頓大學醫學院的白克(Minkyung Baek)說:「我們希望這個新工具將不斷為研究界提供幫助。」◇
-------------------
局勢持續演變
與您見證世界格局重塑
-------------------
🔔下載大紀元App 接收即時新聞通知:
🍎iOS:https://bit.ly/epochhkios
🤖Android:https://bit.ly/epochhkand
📰周末版實體報銷售點👇🏻
http://epochtimeshk.org/stores